Hemoglobin A Structure, Sickle Cell Anemia, and Carbon Monoxide Toxicity I.
Introduction Please leave comments/suggestions or please acknowledge use of this site by visiting our feedback page This exhibit displays molecules in the left part of the screen, and text that addresses structure-function relationships in the right pane. Use the scroll bar to scroll through the text. If using a browser other than Firefox (the recommended browser for this site), be sure to allow popups. In Chrome, you can click on the popup blocker icon in the right part of the address bar. To reset the molecule, use the reset buttons: If you are a practiced user, you can create the illusion of 3D if you turn on stereo mode. In this mode, when you train one eye on one image and the other eye on the other image, you will elicit a centered image that appears truly 3-dimensional. To turn on stereo mode when viewing a scene, return here and use this button . To turn off stereo mode, return here and use this button . To view a hemoglobin molecule in AR (augmented reality) on your phone or notebook, see the directions on the OMM exhibit page and then use your camera to scan this image: |

I. Introduction
The tetrameric protein at left is Hemoglobin A in its oxygenated state, comprising two alpha (α) and two beta (β) globin chains, encoded by an α and β globin gene, respectively. Hemoglobin, the most efficient O2 carrier known, is found in very high concentrations within red blood cells of humans and nearly all other vertebrates. It is also present in a few invertebrates, dissolved directly in their blood. Oxygenated hemoglobin within red blood cells is responsible for ferrying oxygen acquired via gas exchange in the pulmonary capillaries of the lungs to cells throughout the body. The release of oxygen from hemoglobin provides these cells with adequate oxygen for cellular respiration and restores hemoglobin to a deoxygenated state.
The structure of the hemoglobin protein and associated heme cofactors endows it with the remarkable ability to bind and release molecular oxygen (O2) under appropriate conditions. This exhibit is an introduction to hemoglobin structure-function relationships, the pathology of a type of Sickle Cell Disease that some mutations in the β globin gene can produce, and the molecular basis of carbon monoxide toxicity.
Note: if the reader is unfamiliar with the principles of chemical bonds and protein structure, it may be helpful to review the following exhibit: An Introduction to Chemical Bonds and Protein Structure.
II. Tertiary and Quaternary Structure, Heme-mediated O2 Binding/Release, and Cooperativity
(a) Tertiary/Quaternary Structure
1. Each α and β subunit shares a conserved tertiary structure composed mostly of α helices.
2. Two dimers (α1β1 and α2β2) associate to form the tetramer.
3. When fully deoxygenated, i.e. all monomers lacking bound oxygen, multiple salt bridges (ionic bonds) are involved in stabilizing the tetramer's "Tense" state. In the following four ionic bond examples, visualization is clearest if the buttons are clicked in order:
- 3a. The carboxyl terminus group (residue Arg 141) of each α subunit makes a salt bridge with the amino terminus group (residue Val 1) of the other α subunit. One pair of interacting main chain atoms of Val 1 (α1) and Arg 141 (α2) is show here;
- 3b. The side chain of the Arg 141 C-terminal residue of each α subunit forms an ionic bond with the Asp 126 side chain of the other α subunit. One of the pairs of interacting atoms is shown, from Arg 141(α2) and Asp 126 (α1);
- 3c. The side chain of Lys 40 of each α subunit interacts with the carboxy terminus (residue His 146) of the β subunit of the other "dimer"; e.g. Lys 40 of α1 interacts with the C-terminus of His 146 of β2;
- 3d. The side chain of the His 146 C-terminal residue of each β subunit interacts with Asp 94 of the same chain, β2, in the case illustrated here.
4.
These salt bridges, along with other molecular interactions, help stabilize the conformation of the globin tetramer in a deoxygenated state (hemes lack O2). Distances between inonically-bonded atoms are indicated in Angstroms (Å). When molecular oxygen (O2) is bound by one of the monomers, the conformation of the entire tetramer is altered as all monomers rapidly acquire oxygen (hemes carry O2). As the transition occurs, the salt bridges illustrated above are severed: the amino acids involved become separated by distances that essentially break the ionic bonds.** The tetramer has transitioned from a "TENSE" state (deoxygenated) to a "RELAXED" state (oxygenated). To see an animation of this transition, see #12, below).
[** For more information on the relationship between ionic bond strength and atomic distance, see the Ionic Bonds page of the Chemical Bonds and Protein Structure exhibit.]
(b) Heme-mediated O2 binding and release: cooperative interactions between HbA subunits
5. Each of the four Hemoglobin A (HbA) subunits carries a planar heme prosthetic group, detailed here for one of the α subunits. The hemes are pigments that endow hemoglobin with its bright red color and its ability to bind and release oxygen. Each heme consists of a central iron atom held in an aromatic, porphyrin ring. The iron is ionized (Fe+++ when O2 is bound and Fe++ when O2 has been released). The iron is coordinated by four nitrogen atoms in the center of the ring.
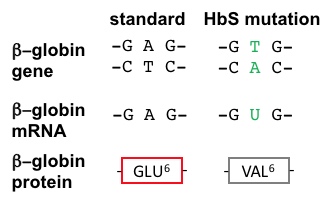
6. The iron is anchored to a globin subunit by a coordinate, or dative, covalent bond to a histidine side chain, termed the F8 or proximal histidine. A coordinate bond occurs when one participating atom contibutes both shared electrons to a covalent bond. The F8 histidine is the 8th residue of the F helix in both α and β globin chains. This coordinate bond links the F helix to the heme, so that changes in the disposition of the iron within the heme plane are transmitted to the protein via the F8 histidine (see below).
7. O2 is reversibly bound by the heme Fe+++ through a coordinate bond. O2 binding results in in an "end-on bent" arrangement: one oxygen atom binds to Fe+++ and the other oxygen protrudes at an angle. Another histidine residue, the E7 or distal histidine, is the 7th residue of the E helix and serves to stabilize bound O2. In a deoxygenated globin, a loosly sequestered H2O molecule takes the place of O2. Although hemoglobin can carry other gases, e.g. carbon dioxide and nitric oxide, these are not bound by the heme, but by side chains elsewhere in the protein.
8. Upon binding O2 in the pulmonary capillaries of the lungs, where the partial pressure of oxygen is high, a globin monomer changes shape due to the movement of the heme Fe from a position slightly "below" the plane of the heme to a position in the heme plane. This draws the F8 (proximal) histidine toward the heme, as can be seen by alternating between an α subunit in its deoxygenated (DEOXY) and oxygenated (OXY) state. The SUPERPOSE freeware program was used to create the OXY< > DEOXY transitions (http://wishart.biology.ualberta.ca/SuperPose/).
9. As the F8 histidine shifts, the entire F helix also moves, slightly altering the conformation of the globin monomer.
10. Thus, an O2 binding event in one globin chain can generate an F8 histidine shift, F helix movement, and a subtle alteration of the subunit. This event can be amplified into a conformational change of the entire hemoglobin tetramer, which facilitates the binding of O2 by other subunits. The amplification is generated because the carboxy ends of the F helices that contain the F8 histidines of each globin subunit lie near the interface of the α1β1 and α2β2 dimers.
11. As hemoglobin reaches tissues with lower O2 partial pressure and O2 is released, an OXY >> DEOXY conformational change of a single subunit is transmitted to the other tetramer subunits, altering tetramer structure and stimulating O2 release by other subunits.
12. The shift in HbA tetramer conformation from an oxygenated to deoxygenated state can be visualized by alternating an overlay of the two stuctures (PDB IDs 2DN1 and 2DN2). The remarkably efficient mechanism by which individual subunits can alter the uptake and delivery by other subunits is known as cooperative binding/release, and O2 can be considered an allosteric regulator of the hemoglobin tetramer. More information on hemoglobin cooperativity is available: see the cooperative binding popup.
III. Hemoglobin and Sickle Cell Anemia
(a) Surface features of standard, HbA tetramers
13. Deoxy-hemoglobin (HbA) is shown at left, with the two alpha and two beta chains highlighted and the hemes lacking bound oxygen.
14. Like other soluble, globular proteins in the aqueous, cellular environment, the HbA tetramer is surrounded by polar solvent water (H2O) molecules (notes: H2O's are not shown to scale and in vivo there would be many more: H2O molecules surrounding the protein).
15. Examining the molecular electrostatic potential of the HbA surface, regions of positive and negative potential are observed. This feature, shared by many soluble, globular proteins, allows weak interactions with polar H2O molecules.
16. Despite the overall polar nature of the HbA surface, there exist some hydrophobic residues at or near this surface.
17. One small cluster of these hydrophobic residues (Phe85, Ala70, Leu88) forms a pocket on the surface of both globin β chains. These pockets play an essential role in the pathology of one form of Sickle Cell Disease, Sickle Cell Anemia (SCA), also known as HbS disease, as discussed below. A second feature that is relevant to understanding the molecular basis of SCA is a glutamic acid residue at position 6 of the β globin chains (GLU6).
18. Note that these hydrophobic pockets are largely, but not completely buried in HbA, shown here for 2 HbA tetamers.
(b) Surface features of mutant, HbS tetramers
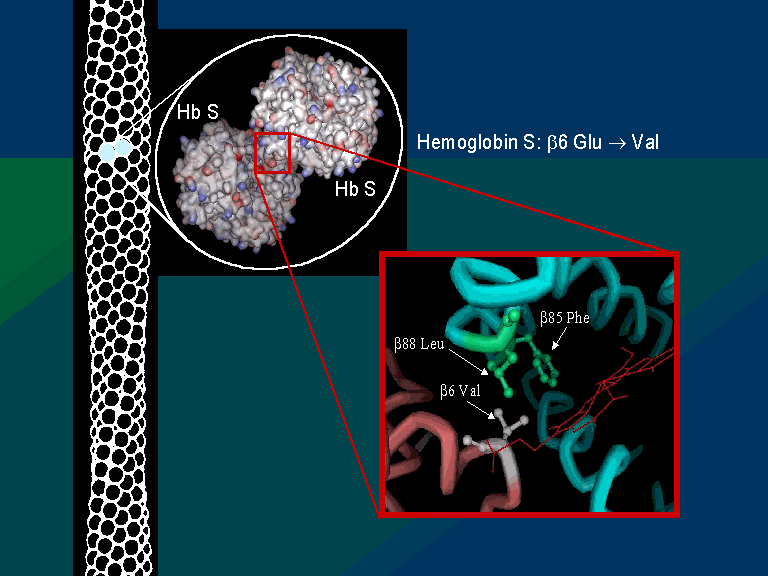
19. The structure at left shows two Hemoglobin S (HbS) tetramers that are "dimerized," representing the beginning of a chain of "polymerized" hemoglobin tetramers that can form long fibers or strands in the red blood cells of Sickle Cell Anemia (SCA) patients. The formation of these strands cause a deformation of the normally disc-shaped red blood cells into crescent-shaped ("sickled") cells that can lodge in small capillaries, thereby blocking blood flow and hence oxygen supply to peripheral tissues (see Figure 1). Note that the two HbS tetramers at left are linked by close contact of a β subunit from each, further elaborated below.
20. SCA Hemoglobin (Hemoglobin S = HbS) is caused by a mutant allele of the β globin gene. The HbS transversion mutation encodes a β subunit of hemoglobin with a hydrophobic valine at position 6 (VAL6) instead of the standard glutamic acid (GLU6): see visualizations above and Figure 2, below. Since GLU6 is a surface amino acid, the HbS mutation results in a β subunit with a hydrophobic VAL sidechain instead of the normal polar GLU sidechain protruding from the surface of the protein, interacting with polar H2O molecules (hydrogens not shown, O's not to scale).
21. Thermodynamically, this is an unfavorable state. Thus, the mutant VAL6 of a β subunit in one hemoglobin tetramer minimizes contact with solvent H2O by packing into a complementary hydrophobic surface pocket of a β chain of another hemoglobin tetramer, mentioned above. The hydrophobic "bonding" of mutant HbS subunits in Hemoglobin S tetramers drives the aggregation of hemoglobin molecules into long, insoluble fibers that deform red blood cells, causing blockages of blood flow in fine capillaries and depriving peripheral tissues of O2 (see Figure 1). This is the molecular basis of SCA. In aggregated fibers, hemoglobin loses its ability to undergo the shifts in tetramer structure associated with cooperative release of oxygen (see cooperativity discussion, above). This exacerbates O2 deprivation in tissues.
Thus, a single, tiny change of one base pair in the β globin gene is responsible for immense amounts of suffering in hundreds of thousands of SCA patients worldwide (~100,000 in the U.S. alone). SCA is only one member of the Sickle Cell Disease family. Other SCDs are caused by different globin mutations.
|
Figure 2. The HbS AT>TA (transversion) mutation in the β globin gene causes an altered mRNA code, leading to a subsitution of a valine for a glutamate at position 6 in the β globin protein. |
22. Note that in unmutated HbA, the GLU6 residue is both sterically and electrostatically incapable of causing hemoglobin tetramer polymerization. This can be seen by modeling a GLU in position 6 of a mutant HbS β subunit in place of a mutant VAL6. The polar, charged GLU sidechain is incompatible with insertion into the hydrophobic surface pocket of a neighboring globin tetramer.
23. Since each HbS beta globin subunit has a VAL6 and a complementary hydrophobic pocket, this affords the opportunity for mutant Hemoglobin S proteins to bind multiple partner tetramers, thus forming multi-stranded fibers that have a semi-helical structure (see Figure 3, below).
|
Figure 3. Assembly of insoluble Hemoglobin S fibers, caused by the polymerization of soluble hemoglobin tetramers due to hydrophobic interactions caused by the GLU6>VAL6 HbS mutation in β globin. Figure from: http://www.unige.ch/sciences/biochimie/Edelstein/sldHbS.htm. Another model of polymerized HbS tetramers (Harrington, et al., 1997) can be seen here. |
IV. Carbon Monoxide Toxicity
24. As stated previously, hemoglobin (Hb) can carry other gases besides oxygen. Carbon dioxide and nitric oxide are examples of gases that can be born by hemoglobin independent of heme cofactors. However, carbon monoxide (CO) binds to the iron of hemoglobin's porphyrin hemes, directly competing with oxygen. This gas is highly poisonous: hemoglobin's binding affinity for CO is ~200 times stronger than its affinity for oxygen! The extreme toxicity of CO lies in its ability to affect the quaternary structure of hemoglobin. CO will bind up to two of the Hb subunits' hemes and allosterically increase the remaining subunits' affinity for O2 (oxygen not shown). However, CO binding decreases the ability of the O2 bearing subunits to release O2, keeping them in a somewhat “locked” state. Thus, even at low partial pressures, CO binding by a single globin chain's heme can prevent oxygen's release in peripheral tissues. Starving cells of oxygen accounts for CO's injurious damage: the gas is potentially lethal even at low concentrations.

V. References
1. Park, S.-Y., Yokoyama, T., Shibayama, N., Shiro, Y., Tame, J.R. (2006). 1.25 a resolution crystal structures of human haemoglobin in the oxy, deoxy and carbonmonoxy forms. J.Mol.Biol. 360: 690-701.
2. Harrington, D.J., Adachi, K., Royer Jr., W.E. (1997). The high resolution crystal structure of deoxyhemoglobin S. J.Mol.Biol. 272: 398-407.
3. Shaanan, B. (1983). Structure of human oxyhaemoglobin at 2.1 A resolution. J.Mol.Biol. 171: 31-59.
4. Berg JM, Tymoczko JL, Stryer L. (2002). Biochemistry. New York: W.H. Freeman.